
EVUSHELD 150 mg 150 mg, solution injectable, boîte de 2 flacons de 1,50 ml
Retiré du marché le : 22/12/2023
Dernière révision : 11/10/2024
Taux de TVA : 2.1%
Laboratoire exploitant : ASTRAZENECA
Source :

Prophylaxie pré-exposition
EVUSHELD est indiqué en prophylaxie pré-exposition de la COVID-19 chez
les adultes et les adolescents âgés de 12 ans et plus pesant au moins
40 kg (voir rubriques Posologie et mode d'administration, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Traitement
EVUSHELD est indiqué pour le traitement des adultes et des adolescents
(âgés de 12 ans et plus pesant au moins 40 kg) atteints de la COVID-19,
qui ne nécessitent pas de supplémentation en oxygène et qui présentent
un risque accru d'évolution vers une forme sévère de la COVID-19 (voir
rubriques Posologie et mode d'administration, Propriétés pharmacodynamiques et Propriétés pharmacocinétiques).
Hypersensibilité aux substances actives ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Hypersensibilité dont l'anaphylaxie
Des réactions graves d'hypersensibilité, dont l'anaphylaxie, ont été rapportées après l'administration d'EVUSHELD (voir rubrique Effets indésirables). En cas d'apparition de signes et symptômes d'une réaction cliniquement significative d'hypersensibilité ou d'anaphylaxie, arrêtez immédiatement l'administration et initier un traitement médicamenteux et/ou des soins de support appropriés.
Evènements cardiovasculaires et/ou thrombo-emboliques
Dans l'étude PROVENT, un plus grand nombre de participants du bras EVUSHELD ont présenté d'évènements indésirables cardiaques ou thromboemboliques graves par rapport à ceux du bras placebo (1,6 % versus 0,9 %). La majorité des participants avaient des facteurs de risque cardiovasculaires et/ou des antécédents de maladie cardiovasculaire pouvant expliquer la survenue de tels évènements.
Une relation causale entre EVUSHELD et ces évènements n'a pas été établie.
Les risques et les bénéfices doivent être évalués avant d'initier EVUSHELD chez les personnes à haut risque d'évènements cardiovasculaires ou thrombo-emboliques. Les patients doivent être informés des signes ou symptômes évocateurs d'un évènement cardiovasculaire (notamment douleur thoracique, dyspnée, malaise, sensation d'étourdissement ou d'évanouissement) et doivent consulter immédiatement un médecin si de tels symptômes surviennent.
Troubles de la coagulation cliniquement significatifs
Comme toutes les autres injections intramusculaires, EVUSHELD doit être administré avec prudence chez les patients qui présentent une thrombopénie ou tout trouble de la coagulation.
Résistance antivirale
Les essais cliniques avec EVUSHELD ont été menés lorsque les variants Alpha, Beta, Gamma et Delta étaient prédominants. Les variants circulants du SARS-CoV-2 peuvent être associés à une résistance aux anticorps monoclonaux tels que le tixagévimab et le cilgavimab. L'activité de neutralisation in-vitro d'EVUSHELD contre les variants du SARS-CoV-2 est présentée dans le tableau 3 (voir rubrique Propriétés pharmacodynamiques).
Les patients ayant reçu EVUSHELD à titre prophylactique doivent être informés du risque potentiel d'infection ultérieure.
La durée de protection pour les variants présentant une diminution de l'activité de neutralisation observée in-vitro est incertaine.
Les patients doivent être informés de consulter rapidement un médecin si des signes ou des symptômes de la COVID-19 apparaissent (les symptômes les plus fréquents incluent la fièvre, la toux, la fatigue et la perte du goût ou de l'odorat ; les symptômes les plus graves incluent la difficulté à respirer ou l'essoufflement, la perte de la parole ou de la mobilité, ou la confusion et la douleur thoracique).
Les décisions concernant l'utilisation d'EVUSHELD pour le traitement de la COVID-19 doivent tenir compte des connaissances sur les caractéristiques des variants circulants du SARS-CoV-2, y compris la prévalence géographique.
Vaccins COVID-19
La prophylaxie pré-exposition avec EVUSHELD n'est pas destinée à être utilisée comme substitut de la vaccination chez les personnes pour lesquelles la vaccination contre la COVID-19 est recommandée.
Excipient à effet notoire :
Ce médicament contient 0,6 mg de polysorbate 80 dans chaque flacon de tixagévimab et dans chaque flacon de cilgavimab. Les polysorbates peuvent provoquer des réactions allergiques.
Résumé du profil de sécurité
Un total de 4 210 participants adultes a reçu 150 mg de tixagévimab et 150 mg de cilgavimab, par injection intramusculaire, dans le cadre du programme de développement de phase III en prophylaxie (incluant PROVENT). Les effets indésirables les plus fréquents (≥ 1%) ont été les réactions au site d'injection (1,6 %) et l'hypersensibilité (1,0 %).
Un total de 452 patients adultes non hospitalisés atteints de formes légères à modérées de COVID-19 a reçu 300 mg de tixagévimab et 300 mg de cilgavimab, par injection intramusculaire, dans l'étude TACKLE. Le profil de sécurité dans son ensemble était similaire à celui rapporté chez les participants qui ont reçu 150 mg de tixagévimab et 150 mg de cilgavimab dans les études de prophylaxie. L'effet indésirable le plus fréquent (≥ 1%) était la réaction au site d'injection (2,4 %).
Tableau récapitulatif des effets indésirables
Les effets indésirables du Tableau 2 sont listés par classe de système d'organe de MedDRA et par fréquence. Les fréquences sont définies de la manière suivante : très fréquent (≥ 1/10) ; fréquent (≥ 1/100 à < 1/10) ; peu fréquent (≥ 1/1 000 à < 1/100) ; rare (≥ 1/10 000 à < 1/1 000) ; très rare (< 1/10 000) et fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Tableau 2 Tableau récapitulatif des effets indésirables
| Classe de système d'organe de MedDRA | Effet indésirable | Fréquencea |
| Affections du système immunitaire | Hypersensibilitéb | Fréquent |
| Anaphylaxiec | Rare | |
| Troubles généraux et anomalies au site d'administration | Réaction liée à l'injectiond | Peu fréquent |
| Blessures, intoxications et complications liées aux procédures | Réaction au site d'injectione | Fréquent |
a Les fréquences sont basées sur les données compilées des études en prophylaxie avec une exposition à 150 mg de tixagévimab et 150 mg de cilgavimab.
b Y compris les termes préférentiels éruption cutanée et urticaire.
c Identifié à partir des rapports post-commercialisation/post-autorisation (voir rubrique Mises en garde spéciales et précautions d'emploi).
d La description des évènements rapportés sous le terme préférentiel Réaction liée à l'injection comprend les céphalées, les frissons et les rougeurs, la gêne ou la douleur à proximité de l'endroit où l'injection a été faite.
e Y compris les termes préférentiels douleur au site d'injection, érythème au site d'injection, prurit au site d'injection, réaction au site d'injection et induration au site d'injection.
Population pédiatrique
Aucune donnée n'est disponible pour les patients pédiatriques de moins de 18 ans (voir rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
INFORMER
le patient de consulter immédiatement un médecin en cas de :
- Apparition
des signes ou symptômes évocateurs d'un évènement cardiovasculaire
(notamment douleur thoracique, dyspnée, malaise, sensation d'étourdissement ou d'évanouissement).
- Apparition des signes ou des symptômes de la COVID-19 comme les
symptômes les plus fréquents : fièvre, toux, fatigue et perte du goût
ou de l'odorat; les symptômes les plus graves incluent la difficulté à
respirer ou l'essoufflement, la perte de la parole ou de la mobilité,
ou la confusion et la douleur thoracique.
Informer les patients ayant reçu le médicament à titre prophylactique du risque potentiel d'infection ultérieure.
INFORMER immédiatement le médecin en cas de :
- symptômes d'un évènement cardiaque : douleur thoracique, essoufflement, sentiment général de gêne, de maladie, ou sensation de malaise, d'étourdissement ou d'évanouissement.
- signes d'une réaction allergique : difficulté à respirer ou à avaler, gonflement du visage, des lèvres, de la langue ou de la gorge, démangeaisons sévères de la peau accompagnées de rougeurs cutanées ou de plaques en relief.
Grossesse
Il n'existe pas ou peu de données sur l'utilisation du tixagévimab et du cilgavimab chez la femme enceinte.
Aucune étude non clinique de toxicité sur la reproduction n'a été réalisée avec le tixagévimab et le cilgavimab (voir rubrique Données de sécurité préclinique). Dans des études de réactivité tissulaire croisée avec tixagévimab et cilgavimab utilisant des tissus humains fœtaux aucune fixation préoccupante sur le plan clinique n'a été détectée. Les anticorps de type immunoglobuline G1 (IgG1) humaine sont connus pour traverser la barrière placentaire ; par conséquent, le tixagévimab et le cilgavimab peuvent être transférés de la mère au fœtus en développement. Le bénéfice potentiel du traitement ou le risque d'un transfert placentaire du tixagévimab et du cilgavimab au fœtus en développement ne sont pas connus.
EVUSHELD ne doit être utilisé pendant la grossesse que si le bénéfice potentiel pour la mère justifie le risque potentiel pour le fœtus.
Allaitement
Il n'y a pas de données disponibles sur l'excrétion du tixagévimab ou du cilgavimab dans le lait maternel, mais il est établi que les IgG maternelles passent dans le lait pendant les premiers jours après la naissance.
Etant donné que le tixagévimab et le cilgavimab ciblent directement la protéine spike du SARS-CoV-2, et compte tenu de la faible absorption systémique après ingestion orale d'anticorps, l'administration d'EVUSHELD peut être envisagée pendant l'allaitement lorsque cela est cliniquement justifié.
Fertilité
Il n'existe aucune donnée sur les effets du tixagévimab et du cilgavimab sur la fertilité humaine. Les effets sur la fertilité masculine et féminine n'ont pas été évalués dans les études animales.
Interactions pharmacocinétiques
Aucune étude d'interaction n'a été réalisée chez l'homme.
EVUSHELD ne devrait pas subir de métabolisme par les enzymes hépatiques ni d'élimination rénale. Le tixagévimab et le cilgavimab ne sont pas excrétés par voie rénale ni métabolisés par les enzymes du cytochrome P450 (CYP) ; par conséquent, des interactions avec des médicaments excrétés par voie rénale ou qui sont des substrats, des inducteurs ou des inhibiteurs des enzymes CYP sont peu probables.
Sur la base d'une modélisation pharmacocinétique, la vaccination après l'administration d'EVUSHELD n'a eu aucun impact cliniquement pertinent sur la clairance d'EVUSHELD.
Sur la base d'une modélisation pharmacocinétique, l'état d'immunodépression n'a pas eu d'impact cliniquement pertinent sur la clairance d'EVUSHELD.
Interactions pharmacodynamiques
Aucune étude d'interaction n'a été réalisée chez l'homme.
EVUSHELD doit être administré par un professionnel de santé.
L'administration doit se faire dans des conditions où la prise en charge des réactions d'hypersensibilité sévères, telles que l'anaphylaxie, est possible. Les patients doivent être surveillés après l'administration selon la pratique médicale locale.
Posologie
Prophylaxie pré-exposition
La dose recommandée chez les adultes et les adolescents âgés de 12 ans et plus pesant au moins 40 kg est de 150 mg de tixagévimab et 150 mg de cilgavimab (Tableau 1), administrés consécutivement sous forme de deux injections intramusculaires distinctes.
Il n'y a pas de données de sécurité et d'efficacité disponibles sur l'administration de doses répétées.
En raison de la diminution de l'activité de neutralisation observée in-vitro, la durée de protection d'EVUSHELD contre certains variants est incertaine (voir rubriques Mises en garde spéciales et précautions d'emploi et Propriétés pharmacodynamiques).
Traitement
La dose recommandée chez les adultes et les adolescents âgés de 12 ans et plus pesant au moins 40 kg est de 300 mg de tixagévimab et 300 mg de cilgavimab (Tableau 1), administrés consécutivement sous forme de deux injections intramusculaires distinctes.
EVUSHELD doit être administré dès que possible après un test virologique positif pour le SARS-CoV- 2 et dans les 7 jours suivant l'apparition des symptômes de la COVID-19 (voir rubrique Propriétés pharmacodynamiques).
Tableau 1 Dose recommandée
| Indication | Dose d'EVUSHELDtixagévimab + cilgavimab | Dose d'anticorps | Nombre de flacons nécessairesa | Volume à prélever du flacon |
| Prophylaxie pré-exposition | 150 mg + 150 mg(1 boîte d'EVUSHELD) | tixagévimab 150 mg | 1 flacon (opercule de couleurgris foncé) | 1,5 mL |
| cilgavimab 150 mg | 1 flacon (opercule de couleurblanche) | 1,5 mL | ||
| Traitement | 300 mg + 300 mg(2 boîtes d'EVUSHELD) | tixagévimab 300 mg | 2 flacons (opercule de couleurgris foncé) | 3,0 mL |
| cilgavimab 300 mg | 2 flacons (opercule de couleurblanche) | 3,0 mL |
a Chaque flacon contient un sur-remplissage pour permettre le prélèvement de 150 mg (1,5 mL)
Patients âgés
Aucun ajustement posologique n'est nécessaire (voir rubrique Propriétés pharmacocinétiques).
Insuffisance rénale
Aucun ajustement posologique n'est nécessaire (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Aucun ajustement posologique n'est nécessaire (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
Aucun ajustement posologique n'est nécessaire chez les adolescents âgés de 12 ans et plus pesant au moins 40 kg (voir rubrique Propriétés pharmacocinétiques). La sécurité et l'efficacité d'EVUSHELD chez les enfants âgés de moins de 12 ans n'ont pas encore été établies. Aucune donnée n'est disponible.
Mode d'administration
Administration par voie intramusculaire.
Le tixagévimab et le cilgavimab doivent être administrés consécutivement sous forme de deux injections intramusculaires à des sites d'injection distincts dans deux muscles différents, de préférence dans les muscles glutéaux.
Chaque boîte contient deux flacons :
tixagévimab solution injectable (opercule de couleur gris foncé) ;
cilgavimab solution injectable (opercule de couleur blanche).
Pour les instructions de manipulation du médicament avant administration, voir rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
Flacon non ouvert
3 ans
Seringues préparées
Les seringues préparées doivent être administrées immédiatement. Si l'administration immédiate n'est pas possible, les durées et conditions de stockage avant et en cours d'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 4 heures à une température comprise entre 2°C et 25°C.
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2°C et 8°C).
Conserver dans l'emballage d'origine à l'abri de la lumière.
Ne pas congeler.
Ne pas secouer.
Pour les conditions de conservation après la ponction initiale du flacon et la préparation des seringues voir la rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'existe pas de traitement spécifique du surdosage avec le tixagévimab et le cilgavimab. Le traitement d'un surdosage doit comprendre des mesures générales de soutien, y compris la surveillance des signes vitaux et l'observation de l'état clinique du patient.
Dans les essais cliniques, des doses intramusculaires allant jusqu'à 300 mg de tixagévimab et 300 mg de cilgavimab et des doses intraveineuses allant jusqu'à 1 500 mg de tixagévimab et 1 500 mg de cilgavimab ont été administrées sans toxicité dose-limitante.
Classe pharmacothérapeutique : Sérums immuns et immunoglobulines, anticorps monoclonaux antiviraux, code ATC : J06BD03
Mécanisme d'action
Le tixagévimab et le cilgavimab sont deux anticorps monoclonaux recombinants humains IgG1?, avec des substitutions d'acides aminés dans les régions Fc, pour prolonger la demi-vie des anticorps et pour réduire leur fonction effectrice et le risque potentiel de renforcement de la maladie par les anticorps (voir rubrique Données de sécurité préclinique). Le tixagévimab et le cilgavimab peuvent simultanément se lier à des régions non chevauchantes du domaine de liaison au récepteur (RBD) de la protéine spike du SARS-CoV-2. Le tixagévimab, le cilgavimab et leur association se lient à la protéine spike avec des constantes de dissociation d'équilibre KD de respectivement 2,76 pM, 13,0 pM et 13,7 pM, bloquant son interaction avec le récepteur ACE2 humain, entraînant un blocage de l'entrée du virus. Le tixagévimab, le cilgavimab et leur association ont bloqué la liaison du RBD au récepteur ACE2 humain, avec des valeurs de CI50 de respectivement 0,32 nM (48 ng/mL), 0,53 nM (80 ng/mL) et 0,43 nM (65 ng/mL).
Activité antivirale
Dans un essai de neutralisation du virus SARS-CoV-2 dans des cellules Vero E6, le tixagévimab, le cilgavimab et leur association ont neutralisé le SARS-CoV-2 (souche USA-WA1/2020) avec des valeurs de CE50 de respectivement 60,7 pM (9 ng/mL), 211,5 pM (32 ng/mL) et 65,9 pM (10 ng/mL). Ces valeurs in-vitro sont corrélées à des concentrations sériques cliniquement efficaces in-vivo de 2,2 µg/mL d'EVUSHELD.
Résistance antivirale
Le SARS-CoV-2 ou le virus de la stomatite vésiculaire recombinant codant pour la protéine spike du SARS-CoV-2 (pseudovirus) ont été passés en série sur des cultures cellulaires en présence du tixagévimab ou du cilgavimab individuellement, ou de l'association de tixagévimab et de cilgavimab. Des variants résistants ont été identifiés après le passage avec le cilgavimab, mais pas avec le tixagévimab ou l'association de tixagévimab et de cilgavimab.
Dans les tests de neutralisation utilisant des pseudovirus SARS-CoV-2 recombinants hébergeant les substitutions individuelles de la protéine spike identifiées dans le SARS-CoV-2 circulant, les variants avec une sensibilité réduite au tixagévimab seul incluaient ceux avec les substitutions F486S (> 600 fois) et F486V (121 à 149 fois) et les variants avec une sensibilité réduite au cilgavimab seul incluaient ceux avec les substitutions R346I (> 200 fois), K444E (> 200 fois), K444Q (> 200 fois) et K444R (> 200 fois).
L'activité neutralisante d'EVUSHELD contre les souches de pseudovirus et/ou de virus vivants du SARS-CoV-2 est présentée dans le tableau 3.
La collecte de données est en cours pour mieux comprendre comment les faibles réductions d'activité observées dans les tests utilisant des SARS-CoV-2 authentiques ou des VLP pseudotypés peuvent être corrélées avec les résultats cliniques.
Tableau 3 Données de neutralisation sur des pseudovirus et sur des SARS-CoV-2 authentiques pour les substitutions des variants du SARS-CoV-2 avec l'association de tixagévimab et de cilgavimab
| Lignée Pango avec substitutions de la protéine spike | Substitutions caractéristiques dans le RBD testées | Facteur de réduction de la sensibilité a | CI50 (ng/mL) | ||
| Pseudovirus b | Virus vivant c | Pseudovirus b | Virus vivant c | ||
| Variants préoccupants | |||||
| B.1.1.7 (Alpha,Royaume-Uni) | N501Y | 1,0-5,2 | 0,5-1,4 | 1,1-9,0 | 4-39,5 |
| B.1.351 (Beta,Afrique du Sud) | K417N:E484K:N501Y | 2,5-5,5 | 0,9-3,8 | 5,6-11,4 | 6,5-256 |
| P.1 (Gamma, Brésil) | K417T:E484K:N501Y | 0,8-1,7 | 0,4-2,0 | 1,8-2,7 | 3,2-8 |
| B.1.617.2(Delta, Inde) | L452R:T478K | 1-1,2 | 0,6-1,0 | 1,9-2,2 | 3-7,5 |
| AY.1/AY.2(Delta [+K417N],Inde) | K417N:L452R:T478K | 1,0 | ND | 1,9 | ND |
| B.1.1.529Omicron, BA.1 (Botswana) | G339D:S371L:S373P: S375F:K417N:N440K: G446S:S477N:T478K: E484A:Q493R:G496S:Q489R:N501Y:Y505H | 132-183 d | 12-30 d | 51-277 d | 147-278 d |
| Omicron BA.1.1 (Multiple pays) | G339D:R346K:S371L: S373P: S375F:K417N: N440K:G446S:S477N: T478K:E484A:Q493R: G496S:Q489R:N501Y:Y505H | 424 d | 176 d | 466 d | 1147 d |
| Omicron BA.2 (Multiple pays) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: S477N: T478K:E484A: Q493R:Q498R:N501Y: Y505H:H655Y:N679K: P681H:N764K | 3,2 | 5,4 | 9,8 | 35 |
| Omicron BA.2.12.1(États-Unis) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: L452Q:S477N:T478K: E484A:Q493R:Q498R: N501Y: Y505H | 5 | ND | 10,7 | ND |
| Lignée Pango avec substitutions de la protéine spike | Substitutions caractéristiques dans le RBD testées | Facteur de réduction de la sensibilité a | CI50 (ng/mL) | ||
| Pseudovirus b | Virus vivant c | Pseudovirus b | Virus vivant c | ||
| Omicron BA.2.75 (Inde) | G339H:S371F:S373P: S375F:T376A:D405N:R 408S:K417N:N440K:G446S:N460K:S477N:T478 K:E484A:Q498R:N501Y:Y505H | 2,4-15 | ND | 1,2-14 | ND |
| Omicron BA.2.75.2(Inde) | BA.2.75: R346T:F486S | > 5 000 e | ND | > 10 000 e | ND |
| Omicron BA.3 (Multiple pays) | G339D:S371F:S373P: S375F:D405N:K417N: N440K:G446S:S477N: T478K:E484A:Q493R:Q498R:N501Y:Y505H | 16 | ND | 34,5 | ND |
| Omicron BA.4 (Multiple pays) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: L452R:S477N:T478K: E484A:F486V:Q498R: N501Y:Y505H | 33-65 d | ND | 65-69,4 d | ND |
| Omicron BA.4.6 (États-Unis) | G339D:R346T:S371F: S373P:S375F:T376A: D405N:R408S:K417N:N 440K:L452R:S477N:T478K:E484A:F486V:Q498R:N501Y:Y505H | > 1 000 e | ND | > 1 000 e | ND |
| Omicron BA.5 (Multiple pays) | G339D:S371F:S373P: S375F:T376A:D405N: R408S:K417N:N440K: L452R:S477N:T478K: E484A:F486V:Q498R: N501Y:Y505H | 33-65 d | 2,8-16 d | 65-69,4 d | 56,6-229 d |
| Omicron BF.7 (États-Unis/Belgique) | BA.4: R346T | > 5 000 e | ND | > 10 000 e | ND |
| Omicron BJ.1 (Multiple pays) | G339H:R346T:L368I: S371F:S373P:S375F: T376A:D405N:R408S: K417N:N440K:V445P: G446S:S477N:T478K: V483A:E484A:F490V: Q493R:Q498R:N501Y:Y505H | 228-424 | ND | 228-848 | ND |
| Lignée Pango avec substitutions de la protéine spike | Substitutions caractéristiques dans le RBD testées | Facteur de réduction de la sensibilité a | CI50 (ng/mL) | ||
| Pseudovirus b | Virus vivant c | Pseudovirus b | Virus vivant c | ||
| Omicron BQ.1 (Nigeria) | BA.5: K444T:N460K | > 2 000 e | ND | > 10 000 e | ND |
| Omicron BQ.1.1 (Multiple pays) | BA.5: R346T:K444T:N460K | > 2 000 e | ND | > 10 000 e | ND |
| Omicron BN.1 (Multiple pays) | G339D:R346T:K356T: S371F:S373P:S375F: D405N:R408S:K417N: N440K:G446S:N460K: S477N:T478K:E484A: F490S:Q493R:Q498R:Y505H | 68 | ND | 61-68 | ND |
| Omicron XBB (Multiple pays) | G339H:R346T:L368I: S371F:S373P:S375F: T376A:D405N:R408S: K417N:N440K:V445P: G446S:N460K:S477N: T478K:E484A:F486S: F490S:Q498R:N501Y:Y505H | > 1 400 e | ND | > 1 600 e | ND |
| XBB.1(Multiple pays) | T19I:del24-26:A27S:V8 3A:G142D:Y144-:H146Q:Q 183E:V213E:G252V:G339H: R346T:L368I:S371F:S37 3P: S375F:T376A:D405N:R 408S:K417N:N440K:V445P: G446S:N460K:S477N:T 478K:E484A:F486S:F490S:Q498R:N501Y:Y505 H:D614G: H655Y:N679K:P681H: N764K:D796Y:Q954H:N969K | > 5 000 e | ND | > 10 000 e | ND |
| Omicron XBB.1.5(Multiple pays) | G339H:R346T:L368I: S371F:S373P:S375F: T376A:D405N:R408S: K417N:N440K:V445P: G446S:N460K:S477N: T478K:E484A:F486S: F490S:Q498R:N501Y:Y505H | > 5 000 e | ND | > 10 000 e | ND |
| Lignée Pango avec substitutions de la protéine spike | Substitutions caractéristiques dans le RBD testées | Facteur de réduction de la sensibilité a | CI50 (ng/mL) | ||
| Pseudovirus b | Virus vivant c | Pseudovirus b | Virus vivant c | ||
| Omicron XBB.1.16(Inde) | T19I:del24-26:A27S:V8 3A:G142D:Y144-:H146Q:E 180V:Q183E:V213E:G252V: G339H:R346T:L368I:S3 71F:S373P:S375F:T376 A:D405N:R408S:K417N:N440K: V445P:G446S:N460K:S 477N:T478R:E484A:F486P:F490S:Q498R:N501 Y:Y505H: D614G:H655Y:N679K:P 681H:N764K:D796Y:Q954H: N969K | > 5 000 e | ND | > 10 000 e | ND |
| Omicron XBB.1.5.10/EG.5 (Multiplepays) | XBB.1.5:F456L | > 5 000 e | ND | 10 000 e | ND |
| Omicron EG.5.1 (Multiple pays) | XBB.1.5:Q52H:F456L | > 5 000 e | ND | 10 000 e | ND |
| Omicron BA.2.86(Multiple pays) | T19I:R21T:L24-:P25-: P26-:A27S:S50L:H69-:V70-:V127F:G142D: Y144-:F157S:R158G: N211-:L212I:V213G:L216F:H 245N:A264D:I332V:G339H: K356T:S371F:S373P:S3 75F:T376A:R403K:D405N: R408S:K417N:N440K: V445H:G446S:N450D: L452W:N460K:S477N: T478K:N481K:V483-:E 484K:F486P:Q498R:N501Y: Y505H:E554K:A570V: D614G:P621S:H655Y:I6 70V:N679K:P681R:N764K:D796Y:S939F:Q954H: N969K:P1143L | > 5 000 e | ND | > 10 000 e | ND |
| Lignée Pango avec substitutions de la protéine spike | Substitutions caractéristiques dans le RBD testées | Facteur de réduction de la sensibilité a | CI50 (ng/mL) | ||
| Pseudovirus b | Virus vivant c | Pseudovirus b | Virus vivant c | ||
| Omicron JN.1 (Multiple pays) | T19I:R21T:L24-:P25-:P2 6-:A27S:S50L:H69-:V70-: V127F:G142D:Y144-:F1 57S:R158G:N211-:L212 I:V213G:L216F:H245N: A264D:I332V:G339H:K 356T:S371F:S373P:S375F:T376A:R403K: D405N:R408S:K417N: N440K:V445H:G446S: N450D:L452W:L455S: N460K:S477N:T478K: N481K:V483-:E484K:F4 86P:Q498R:N501Y:Y505H: E554K:A570V:D614G:P 621S:H655Y:I670V:N679K: P681R:N764K:D796Y:S 939F:Q954H:N969K:P1143L | > 5 000 e | ND | > 10 000 e | ND |
a Gamme de réduction de la puissance in-vitro sur plusieurs ensembles de substitutions concomitantes et/ou laboratoires d'analyse utilisant des tests de grade recherche ; facteur moyen de variation de la moitié de la concentration inhibitrice maximale (CI50) de l'anticorps monoclonal requise pour une réduction de 50 % de l'infection par rapport à la souche sauvage de référence.
b Des pseudovirus exprimant la totalité de la protéine spike du variant du SARS-CoV-2 et des substitutions individuelles caractéristiques de la protéine spike à l'exception de L452Q ont été testés, dont Alpha (+L455F, E484K, F490S, Q493R et/ou S494P), et Delta (+K417N) hébergeant les substitutions additionnelles dans le RBD indiquées qui ne sont plus détectées ou détectées à des niveaux extrêmement faibles au sein de ces lignées.
c Des SARS-CoV-2 authentiques exprimant la totalité de la protéine spike du variant ont été testés, dont Alpha (+E484K ou S494P) hébergeant les substitutions additionnelles dans le RBD indiquées qui ne sont plus détectées ou détectées à des niveaux extrêmement faibles au sein de ces lignées.
d La durée de la protection pour ce variant est incertaine.
e Il est peu probable que le tixagevimab et le cilgavimab soient actifs contre ce variant. ND, non déterminé ; RBD, domaine de liaison au récepteur.
La corrélation entre les données de sensibilité à la neutralisation des pseudovirus ou des SARS-CoV-2 authentiques et le résultat clinique n'est pas connue.
Dans l'étude PROVENT, des données de séquençage recueillies lors des visites de patients étaient disponibles pour 21 participants atteints de COVID-19 symptomatiques (7 ont reçu le tixagévimab et le cilgavimab et 14 ont reçu le placebo). Au niveau d'une fraction allélique ≥ 25 %, les variants préoccupants ou les variants d'intérêt les plus fréquemment observés étaient Alpha (5 évènements au total ; tous dans le bras placebo) et Delta (7 évènements au total ; 6 dans le bras placebo et 1 dans le bras EVUSHELD). Sept séquences de souches ancestrales ont également été observées (3 dans le bras placebo et 4 dans le bras EVUSHELD).
Il est possible que les variants associés à une résistance à l'association de tixagévimab et de cilgavimab puissent présenter une résistance croisée à d'autres anticorps monoclonaux ciblant le RBD du SARS-CoV-2. L'association de tixagévimab et de cilgavimab a maintenu son activité contre les pseudovirus hébergeant les substitutions individuelles de la protéine spike du SARS-CoV-2 (E484D/K/Q, F490S, Q493R, S494P, K417E/N, D420N, K444Q, V445A, Y453F, L455F, N460K/S/T, F486V et Q493K) identifiées dans les variants échappant à la neutralisation d'autres anticorps monoclonaux ciblant le RBD de la protéine spike du SARS-CoV-2.
Dans l'étude TACKLE, des données de séquençage recueillies lors de la visite d'inclusion étaient disponibles pour 749 participants (382 ont reçu le tixagévimab et le cilgavimab et 367 ont reçu le placebo). Au niveau d'une fraction allélique ≥ 25 %, la proportion de participants infectés par des variants préoccupants ou des variants d'intérêt était équilibrée entre les groupes de traitement, incluant les participants infectés avec Alpha, Bêta, Gamma, Delta, Lambda et Mu.
Effets pharmacodynamiques
Dans l'étude PROVENT, après l'administration d'une dose intramusculaire de 150 mg de tixagévimab et de 150 mg de cilgavimab, aux jours 8, 29, 58, 92, 183 et 366, les moyennes géométriques de titrations (MGT) des anticorps neutralisants étaient respectivement de 19, 23, 18, 14, 6 et 3 fois plus élevées que les MGT mesurées dans le plasma de patients convalescents de la COVID-19(MGT = 30,8).
Dans l'étude TACKLE, après l'administration d'une dose intramusculaire unique de 300 mg de tixagévimab et de 300 mg de cilgavimab, les MGT d'anticorps neutralisants observés ont été multipliés par plus de 5 dans le bras EVUSHELD jusqu'au jour 169 par rapport au bras placebo : 16, 14, 22, 18 et 5,3 fois par rapport au bras placebo aux jours 6, 15, 29, 85 et 169, respectivement.
Immunogénicité
Dans l'étude PROVENT, après l'administration d'une dose unique d'EVUSHELD (150 mg de tixagévimab et 150 mg de cilgavimab), des anticorps anti-tixagévimab, anti-cilgavimab et anti- EVUSHELD ont été détectés à la suite du traitement chez 7,6 % (234/3085), 11,3 % (341/3024) et 13,1 % (403/3086) des participants qui ont reçu EVUSHELD et chez qui la présence d'ADA pouvait être évaluée.
Dans l'étude TACKLE, après l'administration d'une dose unique d'EVUSHELD (300 mg de tixagévimab et 300 mg de cilgavimab), des anticorps anti-tixagévimab, anti-cilgavimab et anti- EVUSHELD ont été détectés à la suite du traitement chez 7,3 % (27/372), 12,7 % (46/363) et 14,5 % (54/373) des participants chez qui la présence d'ADA pouvait être évaluée, respectivement.
Aucune preuve d'association entre la présence d'ADA et un impact sur la pharmacocinétique ou la sécurité n'a été observée.
Efficacité clinique
Prophylaxie de la COVID-19
PROVENT était un essai clinique de phase III, randomisé (2/1), en double aveugle, contrôlé versus placebo, évaluant EVUSHELD pour la prophylaxie préexposition de la COVID-19 chez des adultes âgés de ≥ 18 ans. Les participants recrutés étaient des personnes considérées comme à risque accru de réponse inadéquate à l'immunisation active (en raison d'un âge ≥ 60 ans, de la présence de comorbidités, d'une maladie chronique préexistante, d'un état immunodéprimé ou d'une intolérance à la vaccination) ou à risque accru d'infection par le SARS-CoV-2 (en raison de leur situation géographique ou des circonstances au moment du recrutement, par exemple les travailleurs de la santé, y compris le personnel des établissements de soins de longue durée, travaillant dans des environnements industriels à haut risque ou vivant à proximité d'une forte densité, y compris les étudiants dans des dortoirs et des casernes militaires). Les participants ont reçu soit 150 mg de tixagévimab et 150 mg de cilgavimab soit un placebo, administré sous forme de deux injections intramusculaires distinctes. L'étude a exclu les participants avec un antécédent d'infection par le SARS-CoV-2 confirmée par un test de laboratoire ou une positivité pour les anticorps anti-SARS- CoV-2 au moment de la sélection.
Les caractéristiques démographiques à l'inclusion étaient bien équilibrées entre le bras EVUSHELD et le bras placebo. L'âge médian était de 57 ans (avec 24 % de participants âgés de 65 ans et plus et 4 % des participants âgés de 75 ans et plus), 46 % des participants étaient des femmes, 73 % étaient caucasiens, 3 % étaient asiatiques, 17 % étaient noirs/afro-américains et 15 % étaient hispaniques/latino-américains. Sur les 5 197 participants, 78 % présentaient des comorbidités à l'inclusion ou des caractéristiques associées à un risque accru de COVID-19 sévère, notamment une obésité (42 %), un diabète (14 %), une maladie cardiovasculaire (8 %), un cancer, y compris des antécédents de cancer (7 %), une bronchopneumopathie chronique obstructive (5 %), une maladie rénale chronique (5 %), une maladie hépatique chronique (5 %), des médicaments immunosuppresseurs (3 %) et une maladie immunosuppressive (< 1 % ).
L'analyse principale incluait 5 172 participants qui étaient négatifs au SARS-CoV-2 par RT-PCR à l'inclusion, dont 3 441 qui ont reçu EVUSHELD et 1 731 qui ont reçu le placebo. EVUSHELD a réduit de manière significative (valeur de p < 0,001) le risque de maladie symptomatique positive au SARS-CoV-2 par RT-PCR (COVID-19) comparé au placebo (Tableau 4). La durée médiane de suivi après l'administration était de 83 jours.
Tableau 4 Incidence de la COVID-19
| N | Nombred'évènementsa, n (%) | Réduction du risque relatif, % (IC à 95 %) | |
| EVUSHELDb | 3 441 | 8 (0,2 %) | 77 % (46, 90) |
| Placebo | 1 731 | 17 (1,0 %) |
IC = intervalle de confiance, N = nombre de participants dans l'analyse.
a Critère d'évaluation principal, un participant a été défini comme un cas de COVID-19 si son premier cas de maladie symptomatique positive au SARS-CoV-2 par RT-PCR se produisait après l'administration et avant le jour 183.
b 150 mg de tixagévimab et 150 mg de cilgavimab.
L'efficacité était constante dans les sous-groupes prédéfinis, incluant l'âge, le sexe, l'origine ethnique et les comorbidités à l'inclusion ou les caractéristiques associées à un risque accru de COVID-19 sévère.
Parmi les participants qui ont reçu EVUSHELD, il n'y a eu aucun événement de COVID-19 sévère/critique (défini comme une maladie symptomatique positive au SARS-CoV-2 par RT-PCR caractérisée au moins par une pneumonie [fièvre, toux, tachypnée ou dyspnée et infiltrats pulmonaires] ou une hypoxémie [SpO2 < 90 % en air ambiant et/ou détresse respiratoire sévère] et un score de 5 ou plus à l'échelle de progression clinique de l'OMS) contre un événement (0,1 %) parmi les participants qui ont reçu le placebo.
Une analyse post-hoc supplémentaire a été effectuée afin de fournir des données actualisées de tolérance et d'efficacité ; le suivi médian était de 6,5 mois pour les participants des deux bras (EVUSHELD et placebo). La réduction du risque relatif de développer une maladie symptomatique positive au SARS-CoV-2 par RT-PCR était de 83 % (IC à 95 % 66, 91), avec 11/3 441 (0,3 %) évènements dans le groupe recevant EVUSHELD et 31/1 731 (1,8 %) évènements dans le groupe placebo (voir figure 1). Parmi les participants qui ont reçu EVUSHELD, il n'y a eu aucun cas de COVID-19 sévère/critique, contre cinq cas chez les participants qui ont reçu le placebo.
Dans les analyses exploratoires de tous les participants qui ont reçu EVUSHELD ou un placebo, y compris 25 participants qui se sont révélés par la suite avoir été positifs au SARS-CoV-2 par RT-PCR à l'inclusion, la réduction du risque relatif de maladie symptomatique positive au SARS-CoV-2 par RT-PCR était de 78 % (IC à 95 % 59, 88), avec 14/3 460 (0,4 %) évènements dans le bras EVUSHELD et 31/1 737 (1,8 %) évènements dans le bras placebo à un suivi médian de 6,5 mois.
Incidence cumulée (%)
Figure 1 Kaplan Meier: Incidence cumulative de la COVID-19 symptomatique
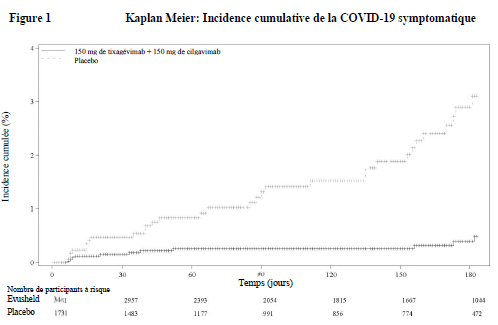
Les variants prédominants du SARS-CoV-2 en circulation pendant la période représentée dans la figure 1 étaient Alpha, Beta, Gamma, Epsilon et Delta. Sur la base de l'incidence des événements du critère d'évaluation principal, la durée d'efficacité était de 6 mois.
Traitement des formes légères à modérées de la COVID-19
TACKLE était un essai clinique de phase III, randomisé (1:1), en double aveugle, contrôlé versus placebo, évaluant EVUSHELD pour le traitement de patients adultes atteints de formes légères à modérées de la COVID-19. L'étude a recruté des individus qui n'avaient pas reçu de vaccin contre la COVID-19, qui n'étaient pas hospitalisés pour un traitement de la COVID-19, et qui présentaient au moins un ou plusieurs symptômes de la COVID-19 d'une gravité au moins légère. Le traitement a été initié dans les 3 jours suivant l'obtention d'un échantillon positif à une infection virale au SARS-CoV- 2 et dans les ≤ 7 jours suivant l'apparition des symptômes de la COVID-19. Les patients ont reçu un traitement standard et soit 300 mg de tixagévimab et 300 mg de cilgavimab (N = 413) soit un placebo (N = 421), administrés sous formes de deux injections intramusculaires distinctes. Les participants ont été stratifiés en fonction du temps écoulé depuis l'apparition des symptômes (≤ 5 jours contre> 5 jours) et du risque de progression vers une forme grave de la COVID-19 (risque élevé versus risque faible).
Les caractéristiques démographiques et celles de la maladie étaient bien équilibrées entre les groupes traitement et placebo. A l'inclusion, l'âge médian était de 46 ans (avec 13 % des sujets âgés de 65 ans et plus), 50 % des sujets étaient des femmes, 62 % étaient caucasiens, 5,6 % étaient asiatiques, 4,0 % étaient noirs et 52 % étaient hispaniques/latino-américains. La majorité des participants (84 %) étaient séronégatifs à l'inclusion, et 90 % étaient considérés comme présentant un risque plus élevé de progression vers une forme sévère de la COVID-19, définis comme étant soit des individus âgés de 65 ans et plus au moment de la randomisation, soit des individus âgés de moins de 65 ans ayant au moins une affection médicale ou un autre facteur les exposant à un risque plus élevé de progressionvers une forme sévère de la COVID-19. Les comorbidités à haut risque comprenaient : l'obésité (IMC ≥ 30) (43 %), le tabagisme (actuel ou ancien) (40 %), l'hypertension (28 %), une maladie pulmonaire chronique ou un asthme modéré à sévère (12 %), le diabète (12 %), une maladie cardiovasculaire (y compris antécédents d'accident vasculaire cérébral) (9 %), un état immunodéprimé (du fait d'une greffe d'organe solide, d'une transfusion ou greffe de moelle osseuse, de déficits immunitaires, du VIH, de l'utilisation de corticostéroïdes, ou de l'utilisation d'autres médicaments immunosuppresseurs) (5 %), un cancer (4 %), une maladie rénale chronique (2 %), ou une maladie hépatique chronique (2 %).
A l'inclusion, 88 % des patients avaient un score de 2 et 12 % avaient un score de 3 sur l'échelle de progression clinique de la COVID-19 selon l'OMS, la durée médiane des symptômes avant traitement était de 5 jours.
Le critère d'évaluation principal de l'efficacité était un critère composite prenant en considération le développement d'une forme sévère de la COVID-19 ou la survenue d'un décès quelle qu'en soit la cause au Jour 29, chez les participants qui ont reçu un traitement dans les 7 jours suivant l'apparition des symptômes et qui n'étaient pas hospitalisés à l'inclusion. La COVID-19 sévère a été définie comme étant caractérisée par une pneumonie (fièvre, toux, tachypnée ou dyspnée, et infiltrats pulmonaires observés sur une radiographie pulmonaire ou une tomodensitométrie pulmonaire) ou par une hypoxémie (SpO2 < 90 % en air ambiant et/ou détresse respiratoire sévère) et un score de 5 ou plus sur l'échelle de progression clinique de l'OMS. EVUSHELD a démontré une réduction statistiquement significative de la COVID-19 sévère ou des décès quelle qu'en soit la cause par rapport au placebo (Tableau 5). Compte tenu de la petite taille de l'échantillon, aucune conclusion ne peut être tirée concernant l'efficacité chez les patients séropositifs.
Tableau 5 Incidence de la COVID-19 sévère ou de décès quelle qu'en soit la cause jusqu'au Jour 29
| Population | Traitement | N | Nombred'événements n (%) | Réduction du risque relatif % (IC à 95 %) | Valeur de pa |
| Patients non hospitalisés recevant une dose≤ 7 jours après l'apparition des symptômes (mFAS) | EVUSHELDb | 407 | 18 (4,4 %) | 50% (15, 71) | p= 0,010 |
| Placebo | 415 | 37 (8,9 %) | |||
| Tous les participants randomisés, y compris les patients hospitalisés et non hospitalisés (FAS) | EVUSHELDb | 446 | 24 (5,4 %) | 42% (5, 64) | p= 0,028 |
| Placebo | 444 | 41 (9,2 %) |
IC = Intervalle de confiance, N= Nombre de participants inclus dans l'analyse, mFAS= Ensemble d'analyse complet modifié, FAS= Ensemble d'analyse complet
Résultats d'un test CMH stratifié en fonction du temps écoulé depuis l'apparition des symptômes (≤ 5 vs. > 5 jours), et du risque de progression vers une forme sévère de COVID-19 (élevé vs. faible).
300 mg de tixagévimab et 300 mg de cilgavimab.
Les données de réponse manquantes n'ont pas été imputées.
La réduction du risque relatif était de 67 % (IC à 95 % de 31, 84) chez les patients non hospitalisés ayant reçu une dose dans les 5 jours suivant l'apparition des symptômes (p= 0,002).
Les résultats du critère composite principal ont été déterminés par l'incidence de COVID-19 sévère. Jusqu'au Jour 29, 7 décès avaient été signalés, 3 dans le bras EVUSHELD et 4 dans le bras placebo. Sur les 7 décès, 2 n'étaient pas liés à la COVID-19. Les deux étaient dans le bras EVUSHELD et ont contribué au critère composite principal.
Population pédiatrique
L'Agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec EVUSHELD dans un ou plusieurs sous-groupes de la population pédiatrique en prophylaxie et en traitement de la COVID-19 (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La pharmacocinétique du tixagévimab et celle du cilgavimab sont comparables, linéaires et dose- proportionnelles entre 150 mg de tixagévimab et 150 mg de cilgavimab et 1 500 mg de tixagévimab et 1 500 mg de cilgavimab après une administration intraveineuse unique. L'analyse pharmacocinétique de population des données de volontaires sains et de patients inclus dans trois études de phase III portant sur le tixagévimab et le cilgavimab en prophylaxie pré-exposition (PROVENT), en prophylaxie post-exposition (STORMCHASER) et dans le traitement de la COVID-19 légère à modérée (TACKLE), ainsi que les données provenant de cinq autres études de phase I et II avec des doses allant de 300 mg (150 mg de tixagévimab et 150 mg de cilgavimab) à 600 mg (300 mg de tixagévimab et 300 mg de cilgavimab) par voie intramusculaire et de 300 mg (150 mg de tixagévimab et 150 mg de cilgavimab) à 3 000 mg (1 500 mg de tixagévimab et 1 500 mg de cilgavimab) par voie intraveineuse, confirme la proportionnalité des doses de tixagévimab, de cilgavimab et d'EVUSHELD.
Absorption
Sur la base de la modélisation pharmacocinétique de la population, après l'administration d'une dose intramusculaire unique de 150 mg de tixagévimab et de 150 mg de cilgavimab, la concentration sérique maximale (Cmax) médiane prédite (intervalle de prédiction [IP] à 90 %) d'EVUSHELD était de 26,9 µg/mL (IP à 90 % : 12,6 ; 53,7), le temps médian pour atteindre la Cmax (Tmax) était de 19 jours (IP à 90 % : 5, 45).
Après l'administration d'une dose intramusculaire unique de 300 mg de tixagévimab et de 300 mg de cilgavimab, la Cmax prédite d'EVUSHELD était de 53,9 µg/mL (IP à 90 % : 25,2 ; 107,3), atteinte après un Tmax médian de 19 jours (IP à 90 % : 5, 46).
La biodisponibilité absolue estimée était de 67,1 % pour EVUSHELD, de 61,5 % pour le tixagévimab et de 65,8 % pour le cilgavimab.
Distribution
Sur la base d'une modélisation pharmacocinétique, le volume central de distribution était de 3,17 L pour le tixagévimab et de 3,52 L pour le cilgavimab. Le volume de distribution périphérique était de 1,77 L pour le tixagévimab et de 1,82 L pour le cilgavimab.
Biotransformation
Le tixagévimab et le cilgavimab devraient être dégradés en petits peptides et composants acides aminés par des voies cataboliques de la même manière que les anticorps IgG endogènes.
Élimination
La clairance (CL) médiane (IC à 95 %) était de 0,050 (0,049 ; 0,052) L/jour pour EVUSHELD, 0,046 (0,044 ; 0,047) L/jour pour le tixagévimab et de 0,052 (0,049 ; 0,054) L/jour pour le cilgavimab avec une variabilité inter-individuelle de respectivement 43 %, 41 % et 44 %. La demi-vie d'élimination terminale médiane (5ème et 95ème percentile) estimée dans la population était de 79 (46 ;101) jours pour EVUSHELD, 81 (49 ; 106) jours pour le tixagévimab et de 78 (49 ; 97) jours pour le cilgavimab.
Après l'administration d'une dose intramusculaire unique de 150 mg de tixagévimab et de 150 mg de cilgavimab, la concentration sérique médiane prédite d'EVUSHELD était de 24,5 µg/mL (IP à 90 % : 11,8 ; 44,8) au jour 29 et de 6,2 µg/mL (IP à 90 % : 1,8 ; 14,7) au jour 183.
Après l'administration d'une dose intramusculaire unique de 300 mg de tixagévimab et de 300 mg de cilgavimab, la concentration sérique médiane prédite d'EVUSHELD était de 49,1 µg/mL (IP à 90 % : 23,6 ; 89,5) au jour 29 et de 12,5 µg/mL (IP à 90 % : 3,6 ; 29,3) au jour 183.
Il n'y avait pas de différence cliniquement pertinente en ce qui concerne la clairance du tixagévimab ou du cilgavimab entre les participants atteints de COVID-19 recrutés dans l'étude TACKLE et ceux recrutés dans les études de prophylaxie.
Populations particulières
Insuffisance rénale
Aucune étude spécifique n'a été conduite pour examiner les effets de
l'insuffisance rénale sur la pharmacocinétique du tixagévimab et du
cilgavimab.
Le tixagévimab et le cilgavimab ne sont pas éliminés sous forme intacte dans les urines, ainsi l'insuffisance rénale ne devrait pas affecter significativement l'exposition au tixagévimab et au cilgavimab. De même, la dialyse ne devrait pas avoir d'impact sur la pharmacocinétique du tixagévimab et du cilgavimab.
Sur la base de l'analyse pharmacocinétique de population, il n'y a pas de différence dans la clairance du tixagévimab et du cilgavimab chez les patients présentant une insuffisance rénale (évaluée par le DFGe initial et la clairance de la créatinine) par rapport aux patients ayant une fonction rénale normale. Dans le modèle pharmacocinétique de population, le nombre de participants atteints d'insuffisance rénale sévère était insuffisant pour tirer des conclusions.
Insuffisance hépatique
Aucune étude spécifique n'a été conduite pour examiner les effets de
l'insuffisance hépatique sur la pharmacocinétique du tixagévimab et du
cilgavimab. L'impact de l'insuffisance hépatique sur la
pharmacocinétique du tixagévimab et du cilgavimab devrait être faible.
Le tixagévimab et le cilgavimab devraient être catabolisés par de multiples tissus via dégradation protéolytique en acides aminés et recyclage en d'autres protéines, par conséquent l'insuffisance hépatique ne devrait pas affecter l'exposition au tixagévimab et au cilgavimab.
Sujets âgés
Parmi les participants à l'analyse combinée de pharmacocinétique, 17,6
% (N = 871) étaient âgés de 65 ans et plus et 3,2 % (N = 156) étaient
âgés de 75 ans et plus. Il n'y a pas de différence cliniquement
significative dans la pharmacocinétique du tixagévimab et du cilgavimab
chez les sujets gériatriques (≥ 65 ans) par rapport aux individus plus
jeunes.
Population pédiatrique
La pharmacocinétique du tixagévimab et du cilgavimab chez les individus âgés de moins de 18 ans n'a pas été évaluée.
En utilisant une modélisation et simulation pharmacocinétique de population, il est attendu que le schéma posologique recommandé entraîne des expositions sériques au tixagévimab et au cilgavimab comparables chez les adolescents âgés de 12 ans et plus et pesant au moins 40 kg à celles observées chez les adultes, puisque des adultes ayant un poids corporel similaire ont été inclus dans les essais cliniques de prophylaxie et de traitement.
Poids corporel élevé
Sur la base d'une analyse pharmacocinétique de population, une
diminution de la concentration sérique maximale d'EVUSHELD et de la
concentration à 6 mois a été observée avec l'augmentation du poids
corporel. La concentration sérique maximale et la concentration à 6
mois chez un adulte pesant 108 kg (percentile 87,5) ont toutes deux été
estimées inférieures d'environ 24 % à celle d'un adulte pesant81 kg (médiane).
Autres populations particulières
Sur la base d'une analyse pharmacocinétique de population, le sexe,
l'âge, la race, l'origine ethnique, les maladies cardiovasculaires, le
diabète et l'immunodépression n'ont eu aucun effet cliniquement
pertinent sur la pharmacocinétique du tixagévimab et du cilgavimab.
EVUSHELD n'a aucune influence ou une influence négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Aucune étude de cancérogenèse, mutagenèse et toxicité sur la reproduction n'a été conduite avec le tixagévimab et le cilgavimab.
Les données non cliniques ne révèlent aucun danger particulier pour l'homme sur la base d'études de liaison tissulaire et d'une étude de toxicité à dose unique chez le singe cynomolgus incluant l'évaluation de la pharmacologie de sécurité et de la tolérance locale.
Risque d'aggravation dépendante des anticorps (ADE) de l'infection
Le potentiel du tixagévimab et du cilgavimab pour médier l'entrée du virus dépendante des anticorps a été évaluée dans des cellules Raji exprimant le FcγRII co-incubées avec des virus recombinants pseudotypés avec la protéine spike du SARS-CoV-2, à des concentrations d'anticorps comprises entre 6,6 nM (1 µg/mL) et 824 pM (125 ng/mL). Le tixagévimab, le cilgavimab et leur association n'ont pas médié l'entrée des pseudovirus dans ces cellules.
Le potentiel d'ADE a également été évalué dans un modèle de primate non humain du SARS-CoV-2 utilisant EVUSHELD. L'administration intravasculaire avant l'inoculation du virus a entraîné une amélioration dose-dépendante de tous les résultats mesurés (ARN viral total dans les poumons ou les muqueuses nasales, taux de virus infectieux dans les poumons basé sur des mesures de TCID50, et lésion pulmonaire et pathologie basées sur des mesures histologiques). Aucun signe de progression de la maladie n'a été observé à aucune des doses évaluées, y compris des doses sous-neutralisantes allant jusqu'à 0,04 mg/kg.
Instructions de manipulation
Ce médicament doit être manipulé par un professionnel de santé en utilisant une technique aseptique afin de garantir la stérilité de chaque dose.
Inspecter visuellement les flacons pour vérifier l'absence de particules et d'un changement de couleur. Le tixagévimab et le cilgavimab sont tous deux des solutions limpides à opalescentes, incolores à légèrement jaunes. Jeter les flacons si la solution est trouble, d'une couleur anormale ou contient des particules visibles. Ne pas secouer les flacons.
Chaque dose de tixagévimab et de cilgavimab est prélevée dans deux seringues distinctes pour être administrée par voie intramusculaire dans deux muscles différents, de préférence dans les muscles glutéaux.
Pour les conditions de conservation des seringues préparées, voir rubrique Durée de conservation.
Toute solution non utilisée doit être jetée.
Elimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament soumis à prescription initiale hospitalière annuelle.
Renouvellement non restreint.
- Indication en prophylaxie pré-exposition de la COVID-19 :
- Agrée aux collectivités en cas de souche de SARS-CoV-2 sensible, dans la prophylaxie pré-exposition de la COVID-19 chez les adultes et adolescents (âgés de 12 ans et plus et pesant au moins 40 kg) ayant un déficit de l'immunité lié à une pathologie ou à des traitements et faiblement ou non répondeurs après un schéma vaccinal complet conformément aux recommandations en vigueur ou non éligibles à la vaccination et qui sont à haut risque de développer une forme sévère de COVID-19.
- Indication non remboursée Séc soc (demande à l'étude).
- Indication en traitement curatif de la COVID-19 :
- Indication non remboursée Séc soc et non agréé Collect au titre de l'AMM à la date du 31 octobre 2023 (demandes à l'étude).
- La prise en charge associée à Evusheld au titre des continuités des traitements est assurée pour une période de trois mois à compter de l'arrêt de la prise en charge précoce de Evusheld au titre de l'article L. 162-16-5-1 du code de la sécurité social, soit à compter du 16 octobre 2023. Seules les continuités des traitements des patients initiés à ce titre sont prises en charge dans les conditions mentionnées à l'article L. 162-16-5-4 du code de la sécurité sociale.
Solution injectable (injection)
Solution limpide à opalescente, incolore à légèrement jaune, avec un pH de 6,0.
Flacon de Tixagévimab
1,5
mL de solution injectable dans un flacon en verre transparent fermé par
un bouchon en élastomère chlorobutyle scellé par une capsule flip-off
en aluminium gris foncé.
Flacon de Cilgavimab
1,5
mL de solution injectable dans un flacon en verre transparent fermé par
un bouchon en élastomère chlorobutyle scellé par une capsule flip-off
en aluminium blanc.
Présentation : Chaque boîte contient 2 flacons : 1 flacon de tixagévimab et 1 flacon de cilgavimab.
Chaque boîte contient deux flacons :
Chaque flacon de tixagévimab contient 150 mg de tixagévimab dans 1,5 mL de solution (100 mg/mL).
Chaque flacon de cilgavimab contient 150 mg de cilgavimab dans 1,5 mL de solution (100 mg/mL).
Le tixagévimab et le cilgavimab sont produits par la technique de l'ADN recombinant sur cellules d'ovaire de hamster chinois (CHO).
Excipient à effet notoire :
Chaque flacon de tixagévimab contient 0,6 mg de polysorbate 80.
Chaque flacon de cilgavimab contient 0,6 mg de polysorbate 80.
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Histidine
Chlorhydrate d'histidine monohydraté
Saccharose
Polysorbate 80 (E 433)
Eau pour préparations injectables